原文
论文链接:https://www.nature.com/articles/s41586-021-03819-2.pdf
用AlphaFold进行非常精确的蛋白质结构的预测(AlphaFold2)
- 发表于 2021 年 7 月 15 日 Nature
- DOI: 10.1038/s41586-021-03819-2
- 自然和科学杂志评选为2021年最重要的科学突破之一
- 2021 年 AI 在科学界最大的突破
前言
- 2020 年 11 月 30 号, deepmind 博客说 AlphaFold 解决了 50 年以来生物学的大挑战
- 2021 年 7 月 15 日华盛顿大学的 Protein Design 团队在发布在 8 月 15 日将在 Science 上发表了一个 RoseTTAFold, 使用深度神经网络进行蛋白质结构的预测
文章结构
- 摘要
- 导论: 一页半
- alphafold2: 两页出头, 模型介绍以及训练细节
- 结果分析: 一页
- 相关工作: 非常短
- 讨论
- 附录方法的细节
- SI: 50页, 详细的解释了每个模型里面的细节
摘要
问题
- 蛋白质对于生命来说是必要的, 了解蛋白质的结构有助于理解蛋白质的功能
- 蛋白质是长的氨基酸序列, 不稳定, 容易卷在一起, 从而形成独特的3d结构, 从而决定了蛋白质的功能
- 预测的困难(蛋白质折叠问题), 只知道很少一部分蛋白质的结构, 实验上通过冷冻方法观察费时费力
现有方法
- AlphaFold1 精度不够, 不在原子的精度
- AlphaFold2 能够达到原子的精度
- AlphaFold2 使用了物理和生物学的知识, 也同样使用了深度学习
应用型的文章
- 问题对于领域来说重不重要
- 结果的好坏, 是不是解决了这个问题
- 找新问题或者开发新模型
导论
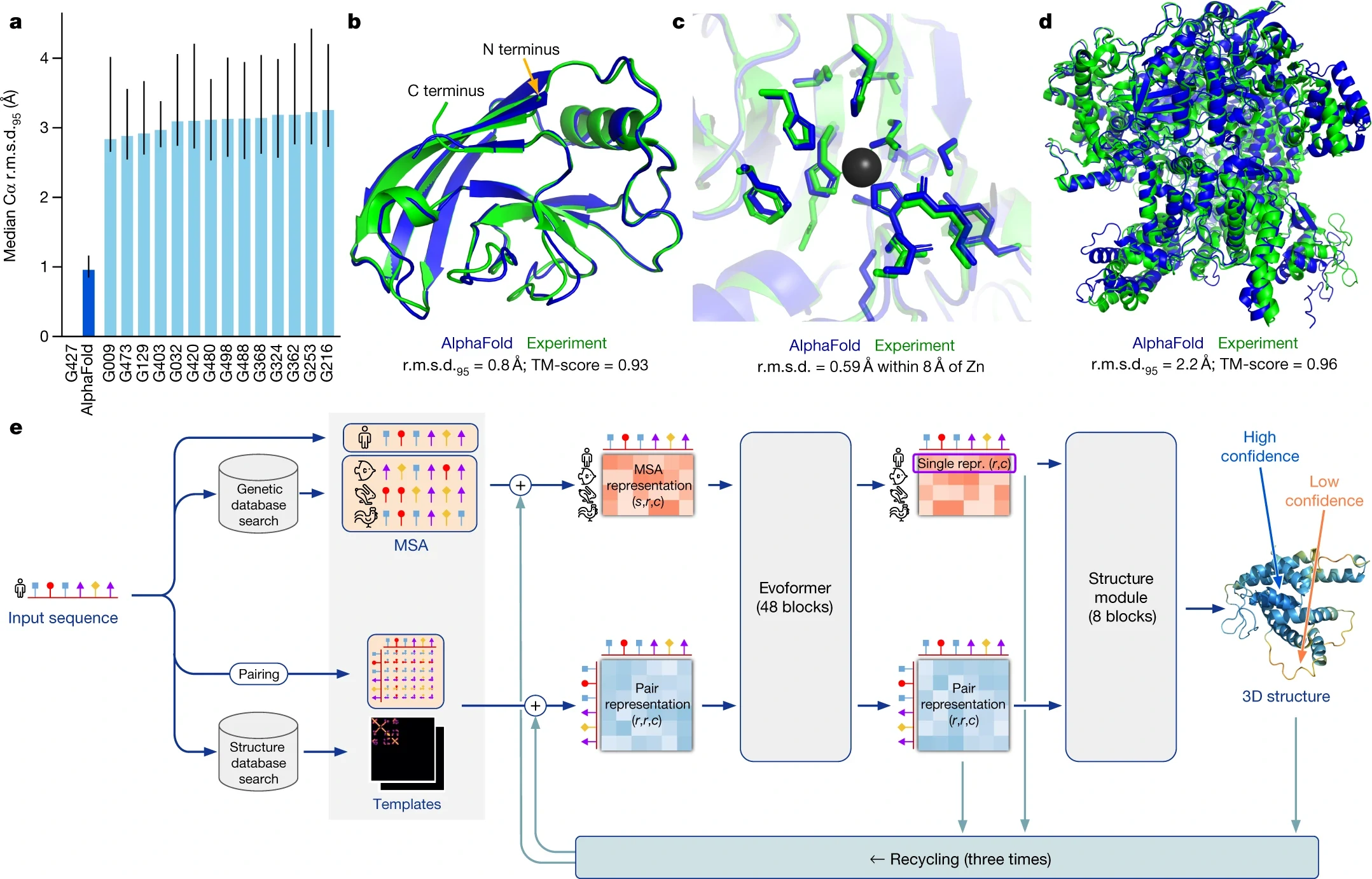
a 图
- x 轴: 参赛队伍
- y 轴: 特定置信区间内, 平均预测的位置和真实的位置在的一个平均的区别(单位是埃)
- AlphaFold 是 1A, 碳原子的大小大概是 1.5A
- 表述结果: 将绝对值变为相对值, 从 1A 到原子精度, 例如图片识别比人类还要好
b, c, d图
- 实验室和计算出来的结果, b 错误率比较小, c 和相对值进行比较, e 复杂的图形
e 图
- 模型大概
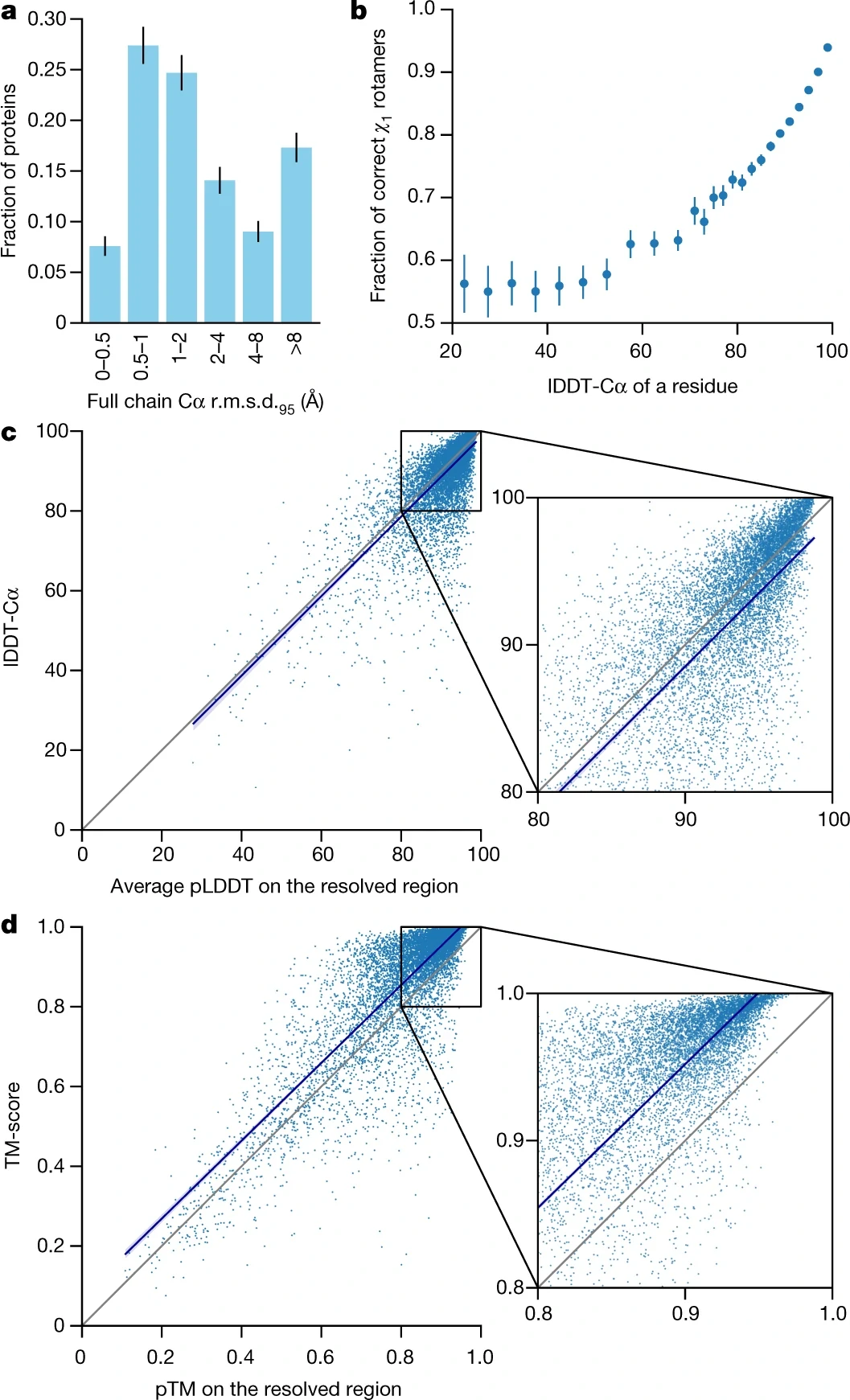
PDB 数据集上 AlphaFold2 的精度
- a 图: 描述数据误差分布结果
模型和训练
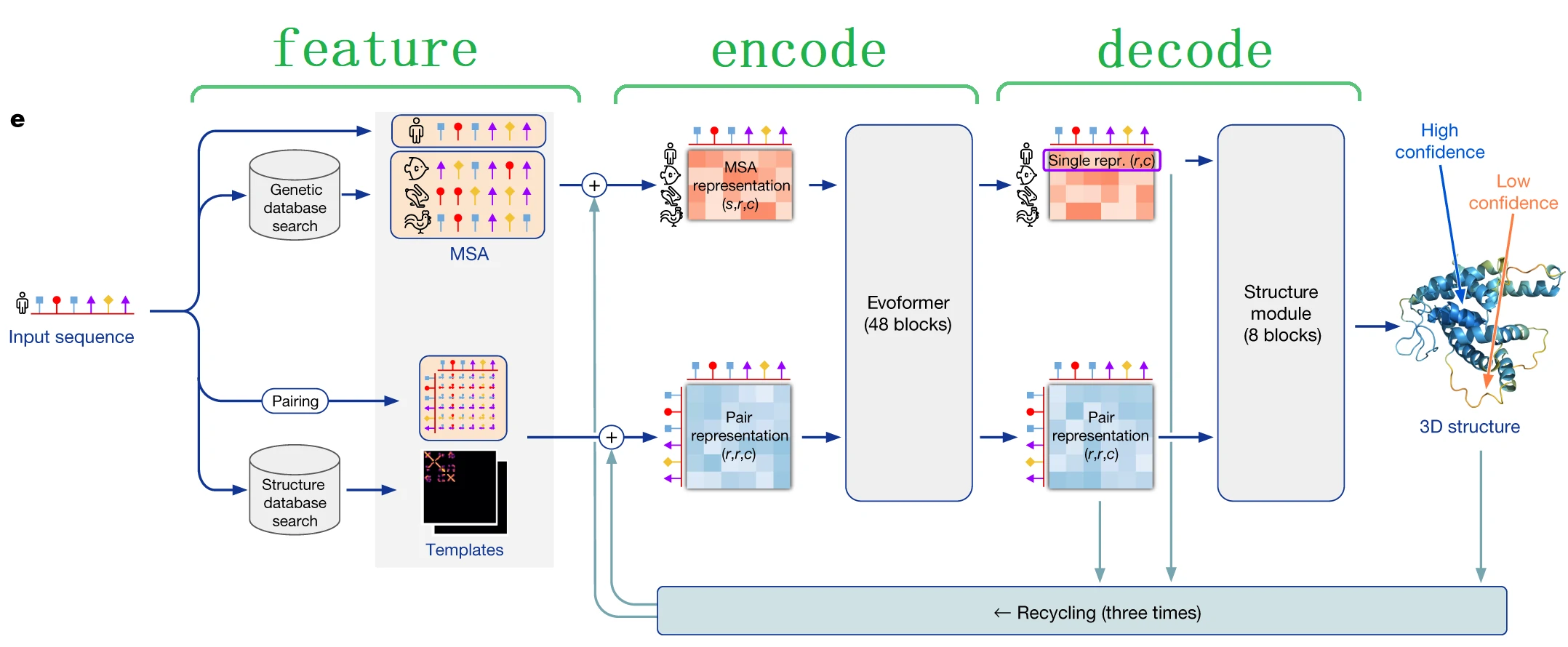
AlphaFold2算法总览图
输入:蛋白质氨基酸的序列
输出:对每个氨基酸序列预测三维空间位置
可以大致分为三部分:
- 第一部分: 抽特征, 主要是两大类特征, 不同序列的特征以及氨基酸之间的特征
- 直接导入神经网络
- 从基因数据库里搜索相似的序列(MSA, 多序列比对), 可以认为是字符串匹配过程
- 氨基酸直接的关系(Pairing), 其中是每个是一对氨基酸之间的关系, 最好是一对氨基酸在三维空间的距离(但是不知道)
- 结构数据库中搜索序列, 真实的氨基酸对之间的信息, 得到很多模板
- 第二部分: 编码器
- 将前面抽取的特征和后面的东西拼起来进入编码器
- 输入是两个三维的张量,MSA 表示(s, r, c),Pair 表示(r, r, c),s 行(第 1 行是要预测的蛋白质, s-1为数据库匹配得到的),r 个氨基酸,长度为 c 的向量表示每个氨基酸
- 进入 Transform 的架构(Evoformer),关系不再是一维的序列而是二维的矩阵,输入的是两个不同的张量,通过 48 个块抽取特征
- 第三部分: 解码器
- 解码器拿到编码器的输出, 目标氨基酸的表示(r, c)和氨基酸对之间的表示(r, r, c)
回收机制
- 将编码器的输出和解码器的输出通过回收机制变成了编码器的输入, 迭代思想或者复制三倍深度更深
- 回收梯度不反传, 经过 56 层(48+8)就能计算梯度
编码器
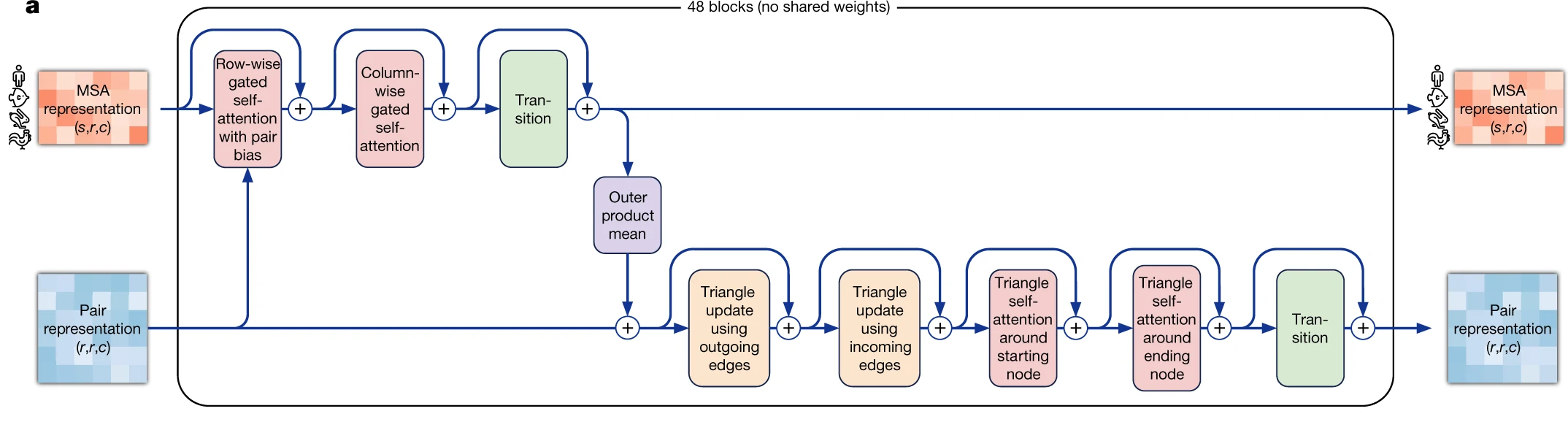
编码器的架构(一块)
- 多头自注意力的模块
- 残差链接 & MLP
- 信息交互: 氨基酸对的信息可以加入序列的建模中, 序列的信息也可以加入到氨基酸对的信息
- 自注意力机制,序列中按行和按列的自注意力机制,氨基酸对中通过物理信息(三角不等式)来设计QKV
- 不共享权重
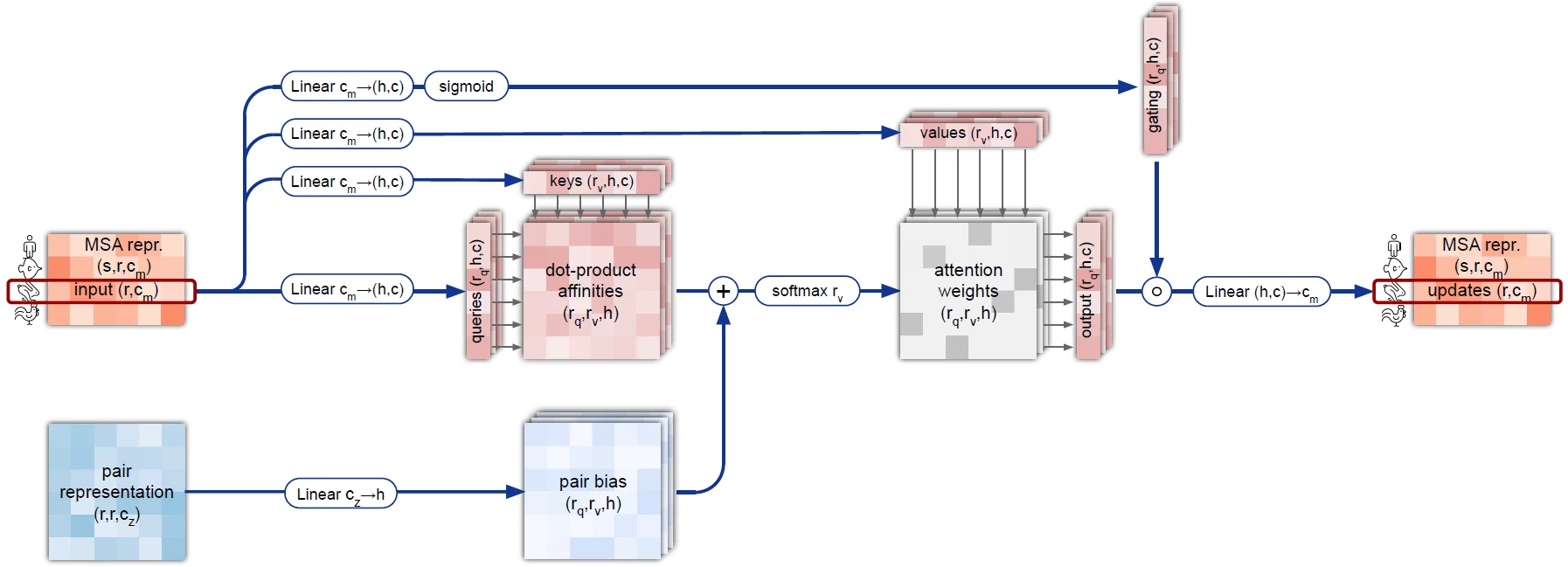

编码器第一个模块(MSA row-wise gated self-attention with pair bias)详细的示意图
- row-wise: 在 MSA 中每次拿出一行做一个序列, 做多头自注意力机制
- gated: 将每个头做一次线性投影以及 sigmoid 计算, 之后和 attention 的输出点乘, 完成门的操作
- pair bias: QK 是 Q 的氨基酸和K的氨基酸的相似度, 这个和 pair 表示有一定的相似的, 将 pair 经过线性投影到 h 维, 从而添加到 QK 中

编码器第二个模块(MSA column-wise gated self-attention)
- 和之前的区别: 按行, 无对信息做偏移加入
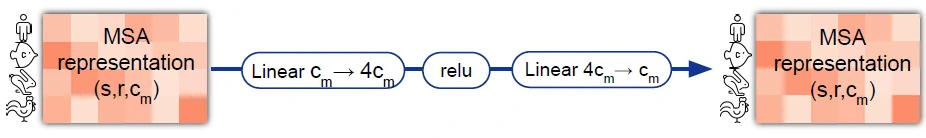
编码器第三个模块MLP(MSA transition layer)
- 自注意力机制主要是混合不同元素之间的信息,做信息的提炼主要还是在 MLP 的模块中
- 将 c 转变为 4c(来自 transform), relu, 之后变为 c
- 线性层的权重对每个元素是共享的
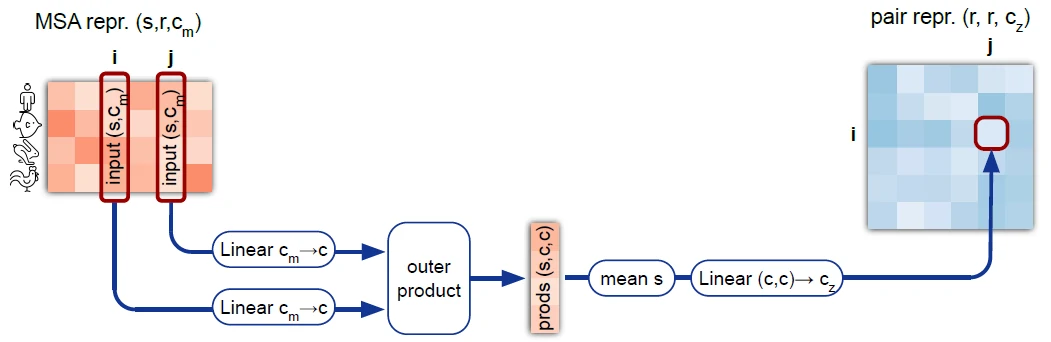
编码器第四个模块(Outer product mean)
- 序列信息融入到氨基酸对的表示中
- 需要将两个矩阵转化为一条向量
- 两个矩阵做外积(s, c, 1) + (s, 1, c) -> (s, c, c)
- 在 s 维度上取均值
- 矩阵拉直, 再投影到 c_z, 加入到对表示
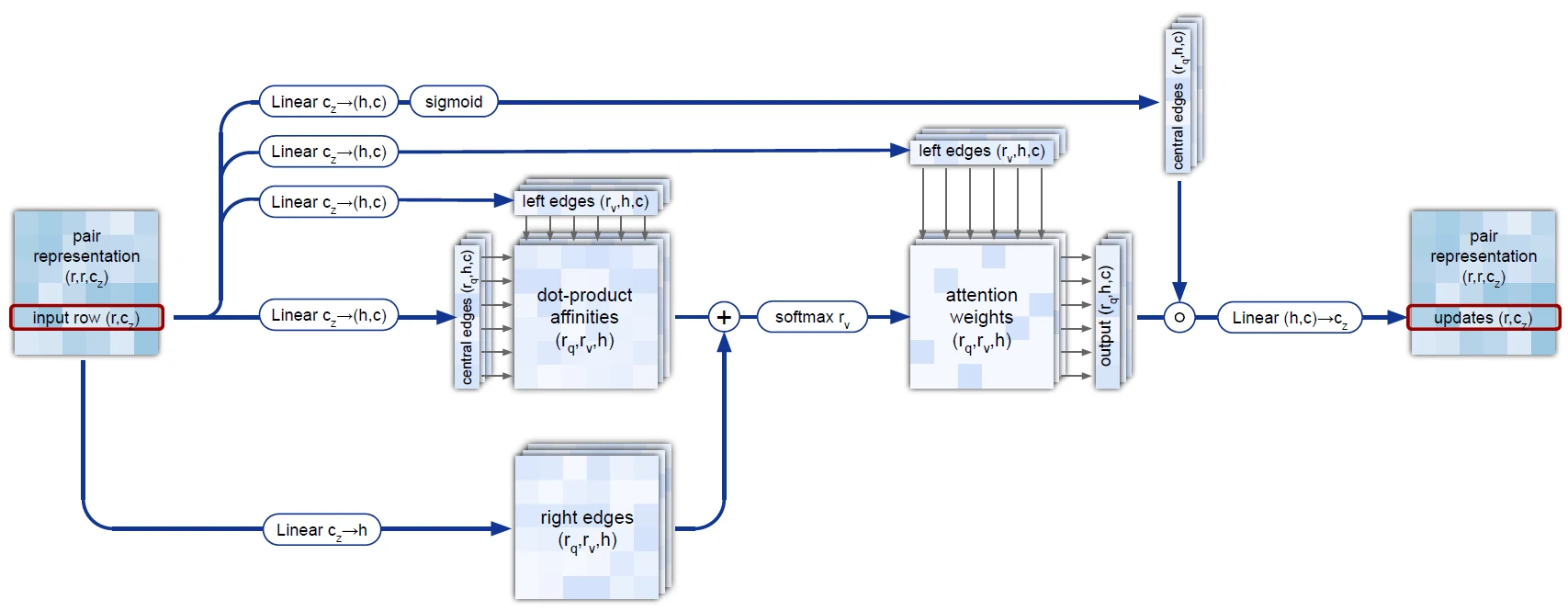
编码器第五个模块(Triangular self-attention around starting node)
- 和 MSA row-wise gated self-attention with pair bias 比较像
- 自注意力机制计算时, 看i, j, k三角的关系, 即 ij + ik < jk(两边和大于第三边)
编码器第五个模块(Triangular self-attention around ending node)
- 和 Triangular self-attention around starting node 的区别从按行做 softmax 转变为按列做 softmax
- 由于先做行后做列, 使得向量是不对称的
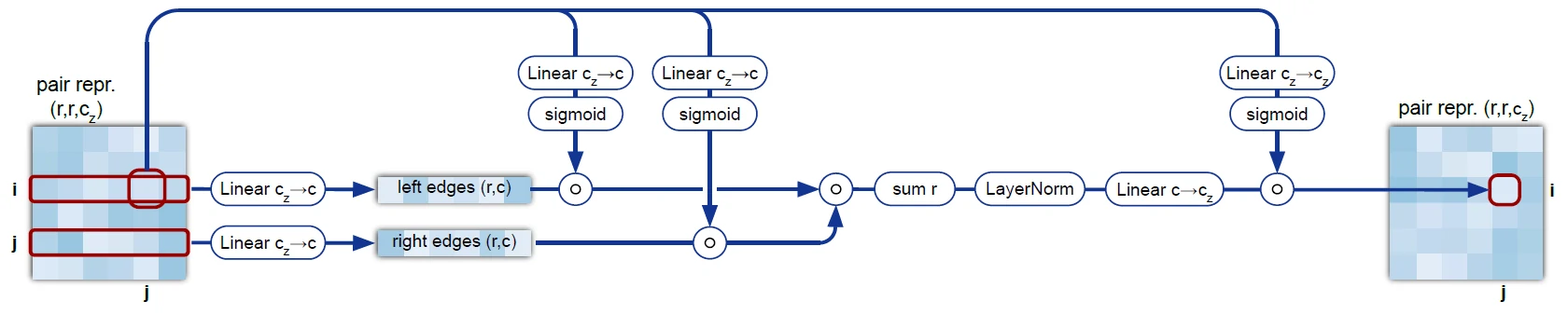
编码器第六个模块(triangular multiplicative update using “outgoing” edges)
- 氨基酸对信息进行交互, 为了替代自注意力模块, 但是没能替换成
- r > s
- ij 之间的信息由所有的 ik 和 jk 信息汇总(遍历 k)
编码器第六个模块(triangular multiplicative update using “incoming” edges)
- 从 ik 和 jk 转变为 ki 和 kj, 对称性的交换
- 氨基酸对之间消息的传递
解码器
蛋白质 3d 结构表达
- 每个原子的空间的绝对值, 不能使用
- 使用相对位置, 下一个氨基酸相对于上一个氨基酸的位置, 使用了欧几里得变换或者说刚体变换
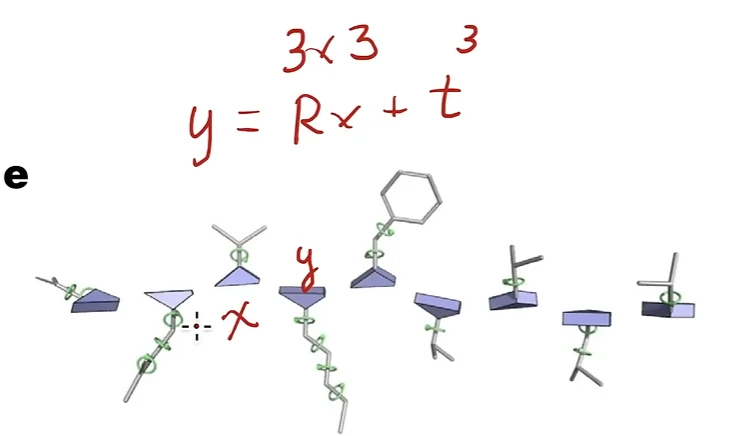
关系氨基酸下面每个原子可以旋转的角度
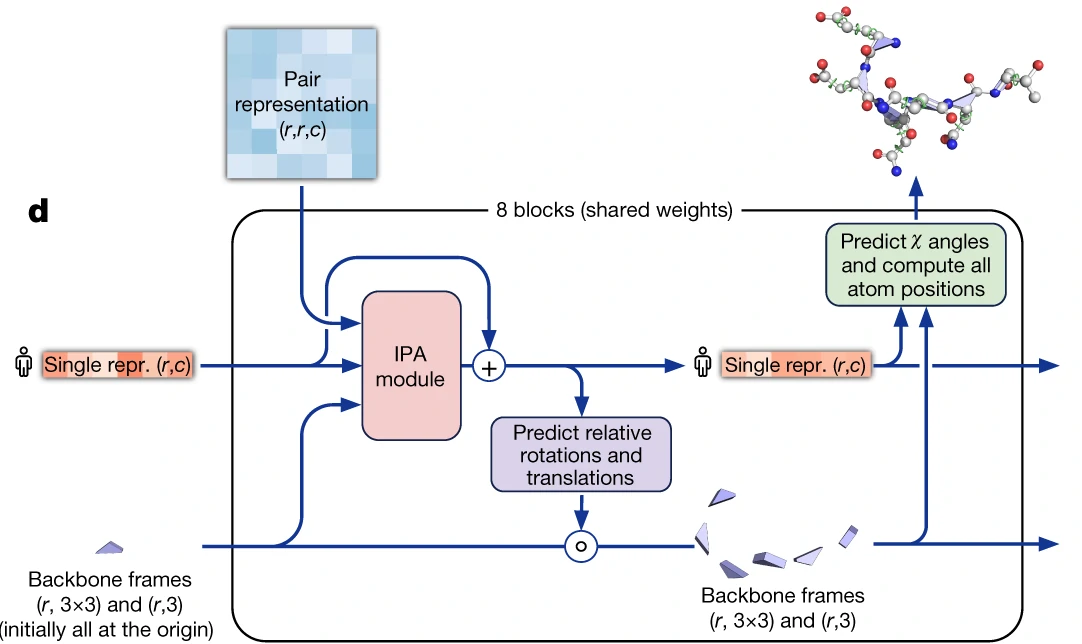
解码器
- 预测主干的旋转和偏移和枝叶的旋转
- IPA 拿到了氨基酸序列以及主干信息
- 不断的调整
- 原始认为在原点
- 共享权重, 像 lstm
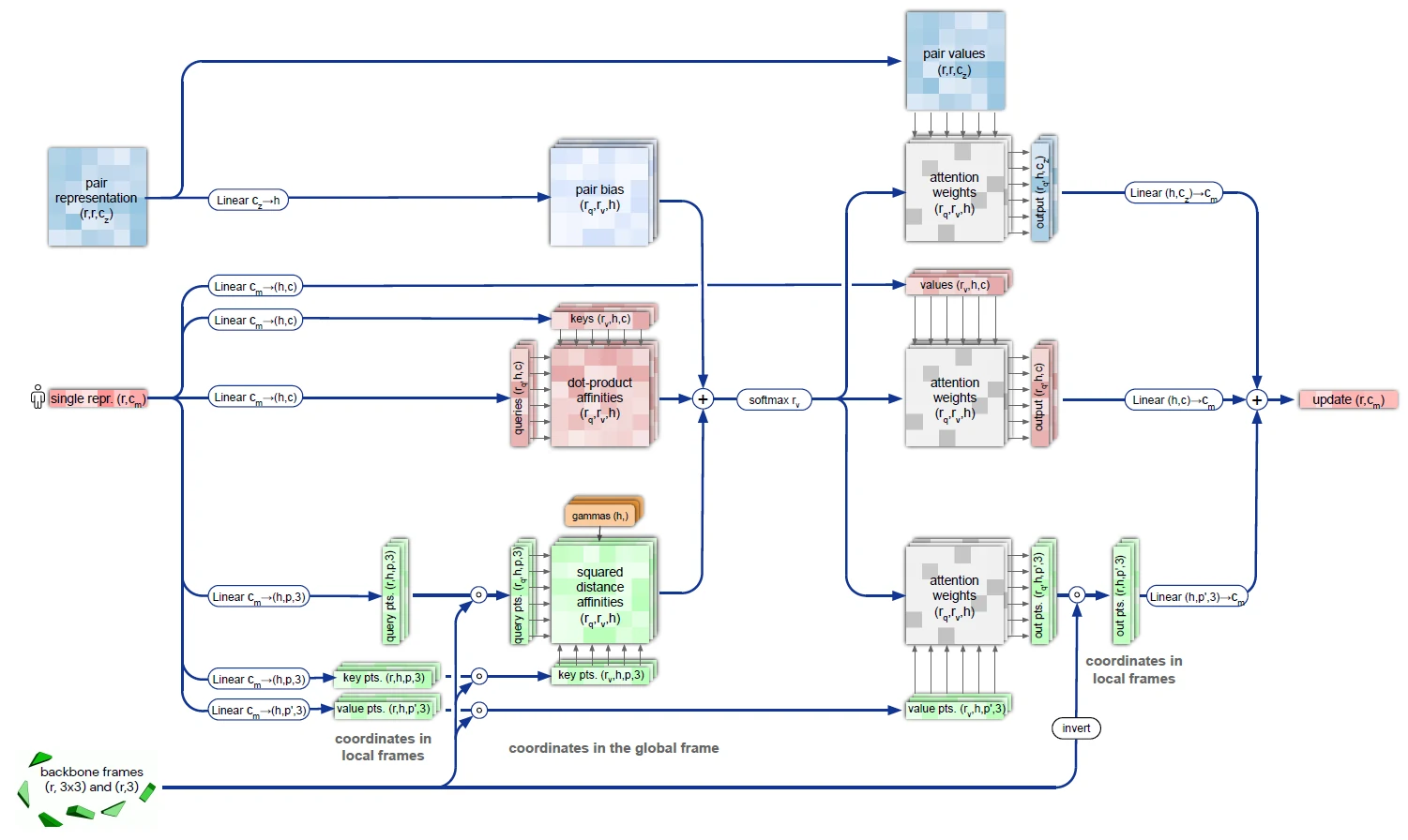

解码器模块-IPA, Invariant Point Attention
- 不动点: 计算距离时不管做什么样的全局变换都不会影响 softmax 里或者输出的值
- 注意力机制, i 和 j 后面两个氨基酸的位置相隔比较远, 那么他们就不应该那么相似
- 解码器中显示的加入了位置的信息
- 经过 IPA 的序列信息理由有位置信息

解码器模块 - Backbone update
- 对第 i 个氨基酸预测它的变换, R 旋转, t 偏移
- 旋转矩阵需要是正交的
- 将 s_i 投影到 6 维里, 通过代数获得有效的矩阵
其他内容
其他的详细内容
- 特征如何抽取, 序列信息有长有短, 搜索到的也有长有短
- 如何将位置信息放入, transform 出了名的对位置信息不敏感
- 回收机制如何执行
损失函数
- 主损失函数: FAPE, 根据预测的变换将蛋白质还原, 对应的原子在真实中和预测出来的位置, 这两距离的相减
使用没有标号的数据
- 使用 noisy student self-distillation
- 核心思想: 先在有标号的数据集上训练一个模型, 然后预测未标号的数据集, 将其中置信的拿出来和有标号的组成新的数据, 重新再训练模型
- 核心关键点: 加噪音, 防止错误标号进入训练级. 加入噪音, 例如大量的数据增强, 甚至把标号改变, 模型能够处理这些不正常的标号
BERT
- 任务: 随机遮住一些氨基酸或者把一些氨基酸做变换, 然后像 BERT 一样去预测这些被遮住的氨基酸
- 训练时加入上述任务, 整个模型对整个序列的键模上更加好一点
训练参数
- 序列长度是 256
- batchsize 是 128
- 128 个 TPUv3 上训练
- 初始训练 7 天, 额外的微调还需 -4 天, 计算两属于中等
- 最大的问题是内存不够, 几百 GB
预测性能(以V100为准)
- 256 个氨基酸, 5min
- 384 个氨基酸, 9min
- 2500 个氨基酸, 18h
结果
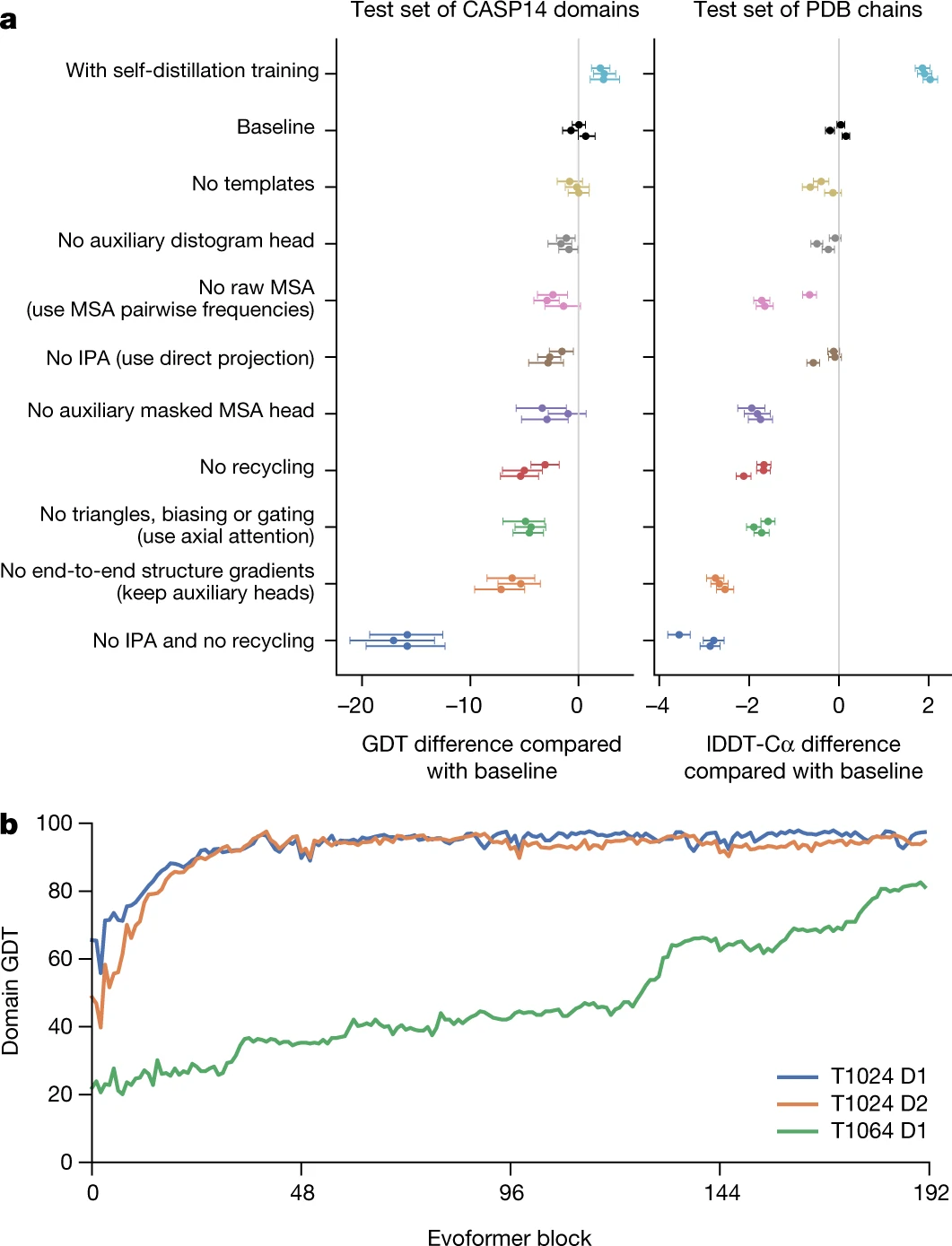
消融实验
- A, casp-14 和 PDB 的结果, 其中灰线是基线, 比 0 大结果好, 比 0 小结果差
- 使用自蒸馏(使用额外的没有标注的数据集进行训练)效果较好
- 去掉数据或者模块的结果
- 我的网络虽然复杂, 但是没有一块能够去掉
编码器的消融(回收), 对于简单的可以只做一次不需要回收, 而对于复杂的 4 次之后仍有上升的趋势
讨论
- 正文比较短, 主要讲问题和模型性能, 对于模型细节比较少
- 对于复杂算法使用相对简单的篇幅进行介绍, 可以学习
- 细节为什么要这么做, 借鉴了很多技术来试
- 提出了:块里面做信息的交互,回收机制,无监督的数据集进行训练